Norrie Disease (ND)
Diese Seite ist auch in deutscher Sprache verfügbar.
Clinical features
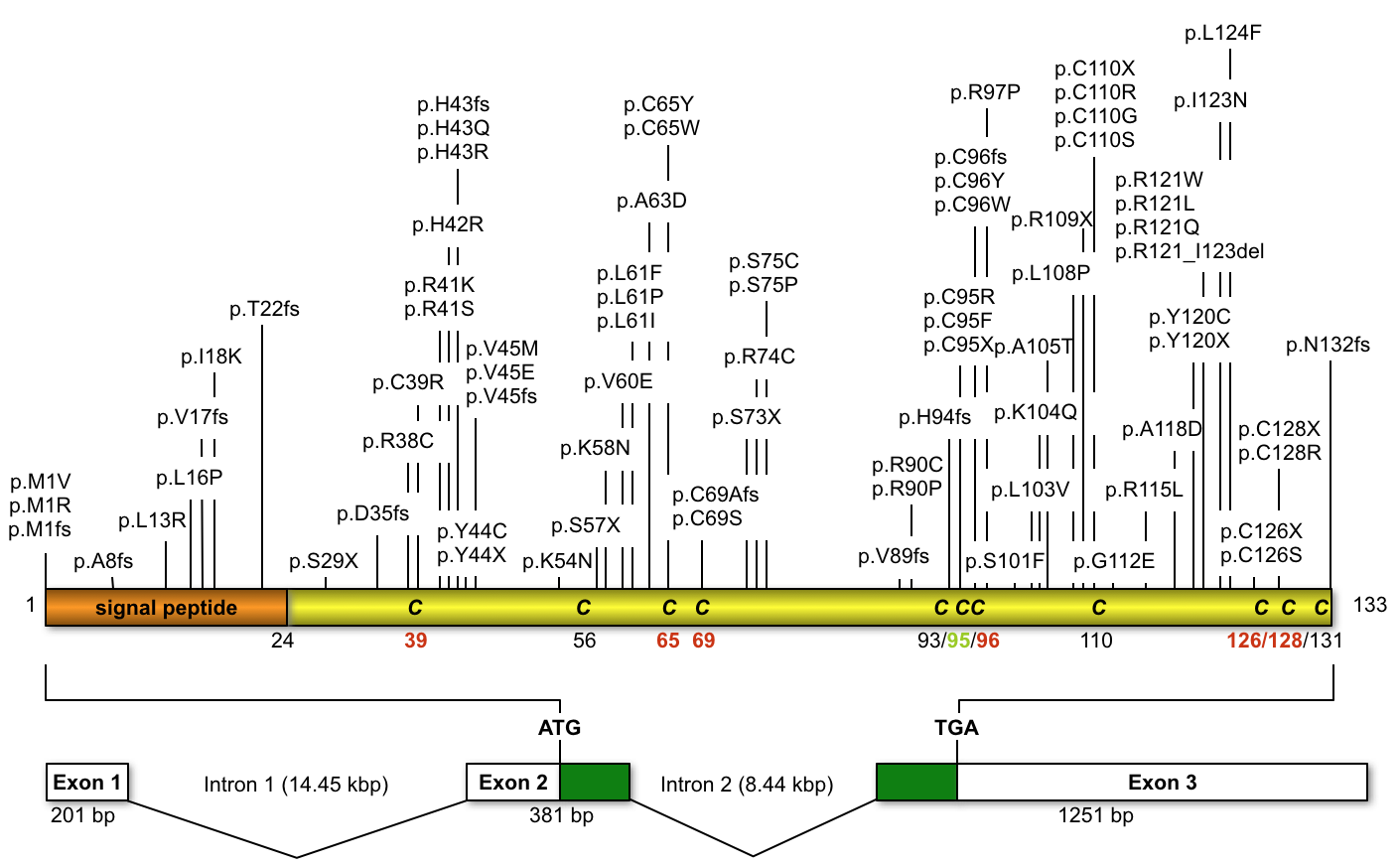
The clinical symptoms of Norrie disease (ND; OMIM 310600), a rare X-linked recessive disorder, comprise congenital blindness, progressive deafness and, in some cases, intellectual disabilities, behavioral difficulties and peripheral vascular diseases (varicose veins, venous insufficiency). ND is caused by mutations in the Norrie disease pseudoglioma (NDP) gene (NDP gene mutations).
Most of the NDP mutations (77%) lead to the classic ND phenotype as described above. However, mutations in this gene have also been found in a variety of other recessive and sporadic vitreoretinal diseases, including exudative vitreoretinopathy (FEVR, OMIM 305390), persistent hyperplastic primary vitreous (PHPV, OMIM 310600) retinopathy of prematurity (ROP, stages 4b and 5) and Coats' disease (OMIM 300216). In a minority of cases (~7%), the clinical spectrum of diseases also includes progressive dyspnea, pulmonary hypertension, sudden death, psychomotor retardation, myoclonic epilepsy as well as immunodeficiency. These more complex phenotypes are frequently associated with large deletions, comprising the NDP gene as well as adjacent loci. High phenotypic variability even in patients from one family carrying the same mutation has been noted, implicating an involvement of modifier genes or other disease-modulating factors.
Reserach in our lab
We have a longstanding interest in Norrie disease as illustrated by gene identification in 1992 by positional cloning and the generation of a mouse model for the disease in 1996. The detailed analysis of the knockout mouse line has revealed striking similarities of the disease phenotype in human patients and affected mice regarding the ocular and audiologic symptoms.
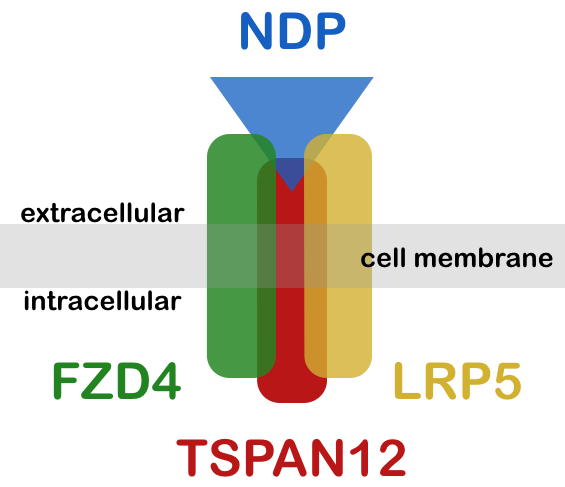
It has been shown that Norrin is a ligand of the Wnt receptor Frizzled-4 (FZD4) and its co-receptors low density lipoprotein receptor-related protein 5 (LRP5) as well as Tetraspanin-12 (TSPAN12). This ligand/receptor complex triggers retinal vascular development, probably by stimulation of endothelial cell proliferation, as we have found recently.
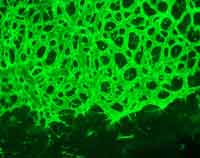
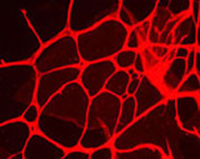
In our lab, we focus on the investigation of the molecular pathophysiology of the disease phenotype, which is similar in different vasoproliferative blindness diseases, especially with regard to abnormalities in the retinal vasculature (angiogenesis).
For this purpose, our group developed a knockout mouse model resembling the human disease in eye and ear. We are analyzing the early development of the disease and its progression by different molecular biological methods, including gene and protein expression (realtime RT-PCR, Western blots, immunohistochemistry). Further, we investigate recombinant protein function in cell culture experiments as well as in vitro assays.
In addition, we are applying human induced pluripotent stem cells (hiPSC’s) in order to develop organoid cultures. The aim of this project is to make use of wildtype or CRISPR/Cas9 edited hiPSC-derived organoids as well as endothelial cells and HUVECs (Human Umbilical Vein Endothelial Cells) to investigate the molecular mechanisms underlying the pathology. Techniques such as quantitative RT-PCR, RNA-sequencing (including single cell sequencing), expression of recombinant wild type and mutant Norrin proteins and subsequent purification, as well as immunocytochemistry are used.
Clinical / genetic testing for ND and FEVR
We also offer clinical / genetic testing for Norrie disease and familial exudative vitreoretinopathies by high throughput sequencing of ATOH7, FZD4, KIF11, LRP5, NDP, TSPAN12 and ZNF408. Norrin, encoded by NDP, is a ligand for the receptor complex consisting of FZD4, LRP5 and TSPAN12. More information on the functional link between the 4 genes is also available on this website at exudative vitreoretinopathies. For details regarding clinical testing please refer to our respective web pages. Genetic Testing
TEAM MEMBERS
Wolfgang Berger (PhD)
Silke Feil
Samuel Koller (PhD)
Kevin Maggi