Familial Exudative Vitreoretinopathies (FEVR)
Our research focusses on the genetic basis of FEVR in sporadic and familial cases by identifying and functionally characterizing mutations in known genes (see below) as well as in novel genes, not yet implicated in FEVR and similar diseases. For this purpose, we use next generation sequencing of retinal gene panels (NGS panels) and whole exome sequencing (WES).
Symptoms and clinical features of FEVR
Familial exudative vitreoretinopathy (FEVR) is characterized by an abnormal vascular development in the peripheral retina of the human eye. Visual problems in patients result from secondary complications caused by retinal ischemia. The avascular retina is present from birth and may progress with long-term complications in adulthood. Expressivity may be asymmetric and highly variable, ranging from mild or asymptomatic to severe (e.g., registered as blind) within the same family. This intrafamilial variability of the disease symptoms might be attributed to genetic and environmental modifiers. The clinical diagnosis is based on fundus fluorescein angiography. FEVR is of particular interest as it involves angiogenic processes.
FEVR can appear as part of a complex phenotype (syndromic FEVR), e.g. in osteoporosis pseudoglioma syndrome (OPPG) or Norrie disease (ND), with an autosomal or X-linked recessive mode of inheritance, respectively. Non-syndromic forms show no extraocular features. The mode of inheritance is autosomal dominant or recessive or X-linked recessive. Rarely, mutations in LRP5 are dominant and TSPAN12 mutations recessive.
Genetic heterogeneity of FEVR
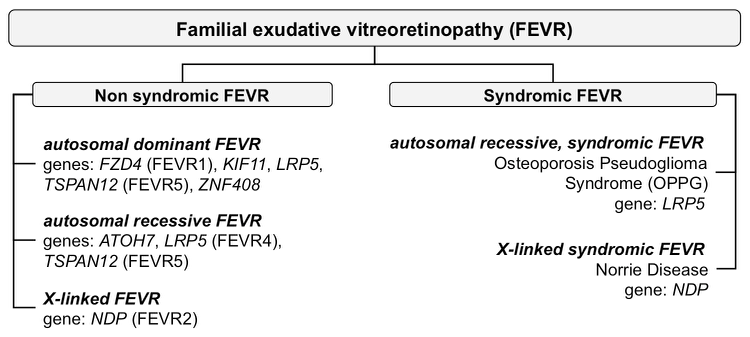
Mutations in eight genes are known to be associated with FEVR: FZD4, encoding the protein frizzled-4; LRP5, encoding low-density lipoprotein receptor-related protein 5; TSPAN12, encoding tetraspanin-12; and NDP, coding for Norrin. More recently, mutations in ATOH7, KIF11, RCBTB1 and ZNF408 were also associated with FEVR. The modes of inheritance of mutations are autosomal dominant (FZD4, KIF11, LRP5, TSPAN12 and ZNF408), autosomal recessive (ATOH7, LRP5, RCBTB1), or X-linked recessive (NDP).
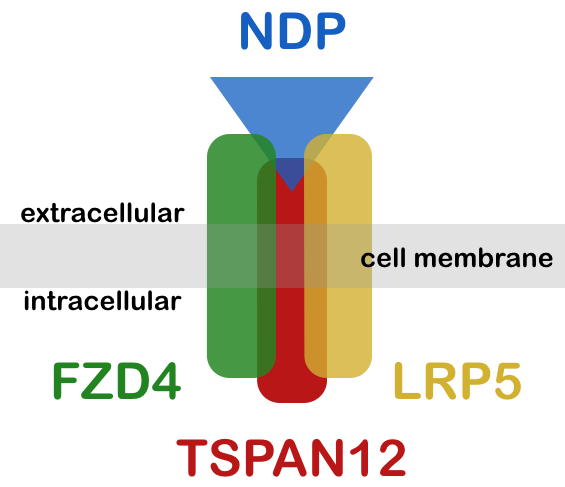
NDP encodes Norrin, a ligand of the receptor and co-receptors FZD4, LRP5 and TSPAN12. This ligand-receptor complex is known to trigger retinal angiogenesis. This mechanism is highly relevant not only to many other human diseases with abnormal vascular development but also for therapeutic intervention.
Table: Genes and numbers of mutations associated with FEVR.
| Gene | Number of Exons | Protein | Number of Amino Acids | Number of Mutations* |
|---|---|---|---|---|
| FZD4 | 2 | Frizzled-4 | 537 | 131 |
| LRP5 | 23 | LRP51 | 1615 | 130 |
| TSPAN12 | 8 | Tetraspanin-12 | 305 | 81 |
| NDP | 3 | Norrin | 133 | 39 |
| KIF11 | 22 | KIF112 | 1056 | 26 |
| ZNF408 | 5 | ZNF4083 | 720 | 18 |
| ATOH7 | 1 | ATOH74 | 152 | 2 |
| RCBTB1 | 13 | RCBTB15 | 531 | 2 |
*Numbers from Human Gene Mutation Database (HGMD), September 2021
1Low-density lipoprotein receptor-related protein 5
2Kinesin family member 11
3Zinc finger protein 408
4Atonal homolog 7
5RCC1 and BTB domain containing protein 1
Clinical variability of symptoms
The severity of FEVR varies tremendously and is unpredictable, even within the same family and even between the eyes of the same patient. There are overlapping clinical features or symptoms due to mutations in the four genes.
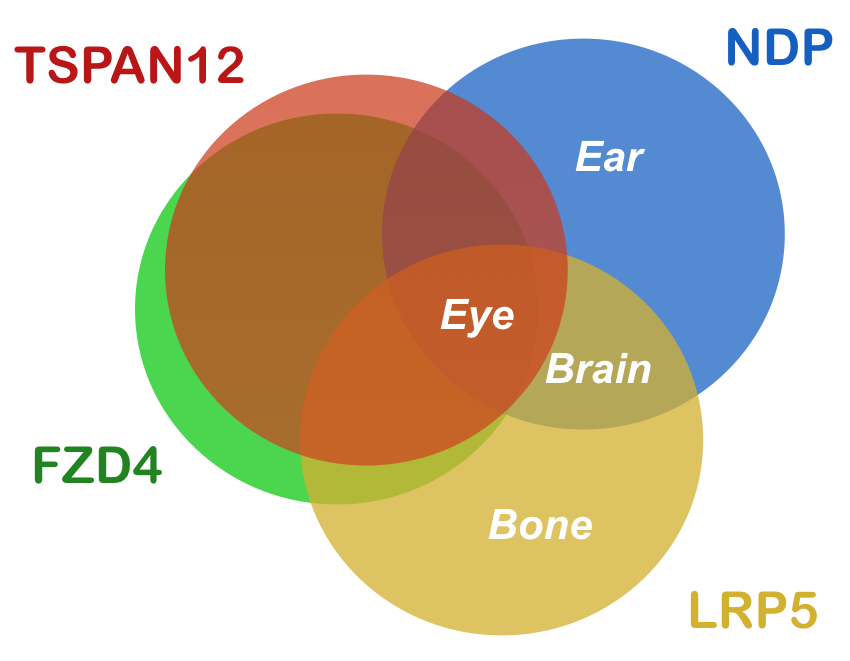
Mutations in NDP may give rise to blindness, deafness and intellectual disability and thus affect ocular, auditory and central nervous tissues. In contrast, mutations in FZD4 and TSPAN12 do exclusively result in ocular symptoms while LRP5 mutations also may affect bones and brain. Also, the mode of inheritance can be variable. FZD4 and TSPAN12 carry dominant mutations in the vast majority of cases, while LRP5 mutations are usually autosomal recessive or dominant and NDP mutations X-linked recessive. In the latter case, only males are affected and female carriers have no symptoms, with only very few exceptions.
Treatment
Treatment of symptoms is of limited success and includes laser photocoagulation to induce the regression of neovascular growth caused by ischemia, and surgical retinal reattachment.
TEAM MEMBERS
Wolfgang Berger (PhD)
Silke Feil
Samuel Koller (PhD)