Optic Nerve Hypoplasia
General information
Congenital visual impairment can be caused by optic nerve hypoplasia (ONH; OMIM #165550), a non-progressive condition where the optic nerves and associated structures, including the optic chiasm, are underdeveloped. In most extreme cases of ONH, the optic nerves are completely missing, and the condition is referred to as optic nerve aplasia. The cause of this anomaly is a significant reduction of retinal ganglion cells in the embryonic retina, resulting in an inadequate optic nerve formation. The prevalence of ONH in European countries has been estimated to lie between 2.4-17.3 cases per 100’000 children. Although it is a rather rare disorder, it has been recognized as a common cause of childhood blindness. The majority of cases are bilateral, albeit asymmetrical affliction of the optic nerves does occur. It is estimated that about 80 % of patients with bilateral ONH are legally blind.
Clinical features
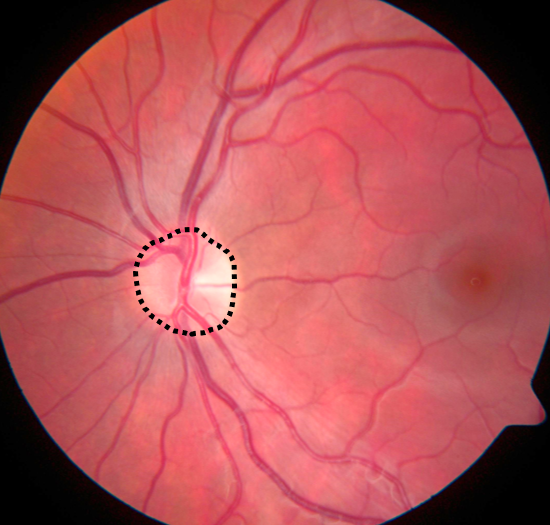
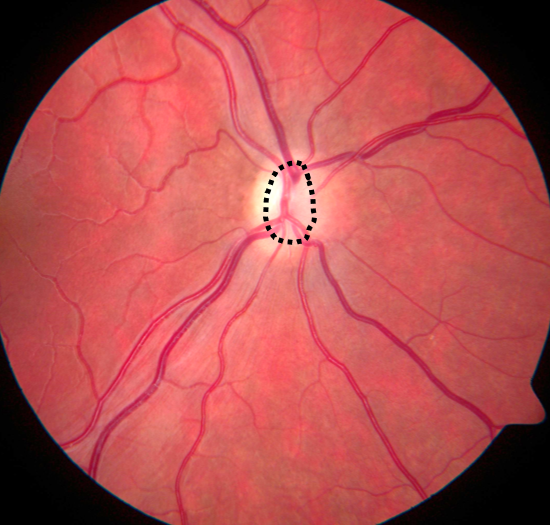
ONH is diagnosed by fundoscopic observation of small optic discs in relation to the size of the overlying central retinal vessels. The findings may be accompanied by irregularities in the optic vasculature, such as abnormal twisting or unusually straight vessels with decreased branching, and sometimes an area of altered pigmentation surrounding the hypoplastic disc. ONH may be confirmed by high-resolution magnetic resonance imaging (MRI), which provides a more reliable assessment of the intracranial part of the optic nerves.
Etiology
Research on the underlying causes of ONH has been mostly focused on environmental risk factors, as cases of ONH are usually sporadic. Several prenatal exposure factors, such as smoking, alcohol, and drugs, have been suggested as causative of ONH, however most of the described associations have been based on single-case reports. The two most consistent risk factors for ONH so far are reported to be young maternal age and primiparity, which may be surrogate factors for poor health and/or risk behavior during pregnancy.
As of today, an increasing amount of genetic mutations are being associated with sporadic cases of ONH. These have been described mostly in early transcription factors involved in retinal and central nervous development, including HESX1 (OMIM *601802), PAX6(OMIM *607108), SOX2 (OMIM *184429), NR2F1 (OMIM *132890), OTX2 (OMIM *600037), VAX1 (OMIM *604294) and ATOH7 (OMIM *609875). Other categories of associated genes include chromatin remodeling, α-dystroglycan glycosylation, cytoskeleton scaffolding, RNA splicing, MAPK signaling pathway, aldehyde dehydrogenases and triglyceride hydrolases. Although there is growing evidence for genetic causes of ONH, familial cases showing this developmental abnormality remain very rare.
Our research
We have reported a Swiss family where two siblings presented bilateral ONH, as well as foveal hypoplasia and minor disturbances in retinal vascularization (Atac et al., 2020). Whole-exome sequencing revealed compound heterozygous mutations in atonal bHLH transcription factor 7 (ATOH7), causing a nonsynonymous substitution in an evolutionary conserved amino acid position located in the dimerizing helix-loop-helix domain of this retinal transcription factor. Further functional characterization of the mutant protein revealed complete loss of transcriptional activity. We are currently investigating the role of ATOH7 in human retinal development utilizing retinal organoids derived from human induced pluripotent stem cells.
TEAM MEMBERS
Wolfgang Berger (PhD)
Silke Feil
David Grubich Atac (MD/PhD)
Samuel Koller (PhD)
COLLABORATORS
Christina Gerth-Kahlert (MD), Dept. Ophthalmology, University Hospital Zurich