Leber Congenital Amaurosis (LCA)
Symptoms and clinical features
Leber congenital amaurosis (LCA) is the most severe form of retinal dystrophies in terms of visual loss and has a very early age of onset (<1 year of age). The clinical features include congenital or early onset loss of vision, sensory nystagmus and no light response upon electrophysiology (ERG). The frequency of the disease is about 1 in 50,000.
Genetic heterogeneity of LCA
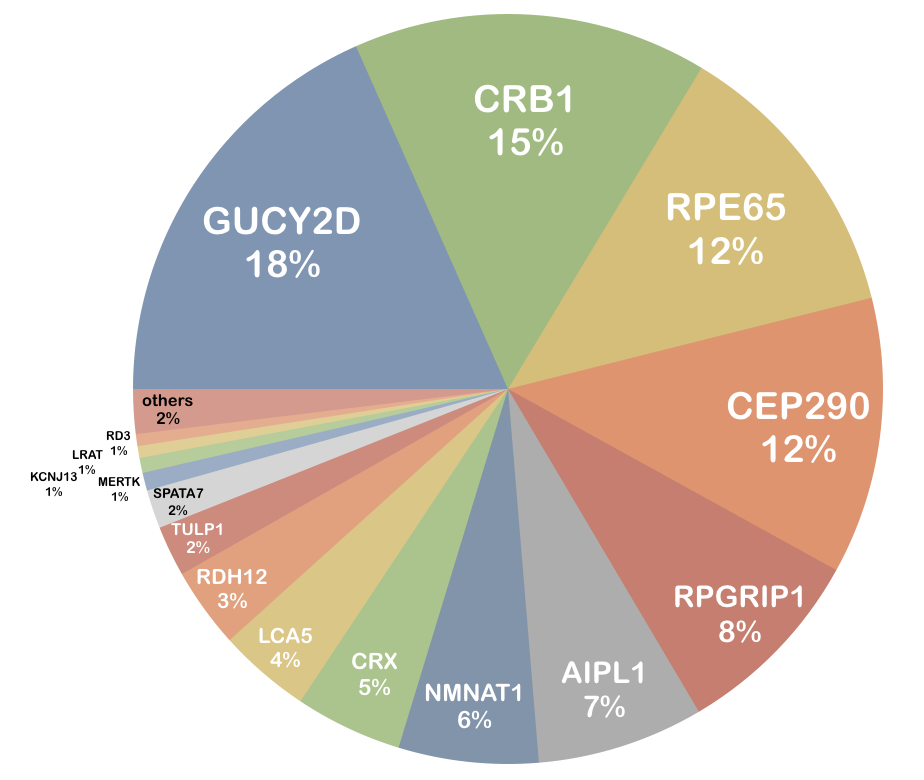
The genetic heterogeneity of this disease is considerable. More than 20 genes were identified to carry mutations in patients affected by this disease. Most of them were detected in GUCY2D (n=143, as of September 2015 in HGMD Professional). Eighteen percent of all known LCA-associated mutations were found in this gene, followed by mutations in CRB1 (15%), RPE65 (12%) and CEP290 (12%). The contribution of mutations in other genes is below 10% for each individual gene.
The mode of inheritance for most of the mutations is autosomal recessive. Dominant inheritance is discussed for sequence alterations in CRX and IMPDH1. The different gene products are involved in many cellular functions including photoreceptor development and differentiation, phototransduction, vitamin A metabolism and others. Because of the recessive nature of most mutations, genetic homozygosity mapping is a particularly successful approach in order to identify the underlying molecular basis of the disease within families.
Treatment
There is no specific treatment for this retinal degeneration except for one form which is caused by mutations in the RPE65 gene. Gene replacement therapy by using adeno-associated virus (AAV) as vector was performed and successful in a group of patients with RPE65-associated LCA. This was a major breakthrough towards treatment of retinal diseases in general and was reported first in 2008 by a group from the Institute of Ophthalmology at University College London (Bainbridge et al. 2008). A second study also indicated successful treatment by RPE65 gene replacement with AAV (Hauswirth et al. 2008).
Our research
Our research focuses on the genetic basis of LCA in sporadic and familial cases by identifying and functionally characterizing mutations in known genes (see link below) as well as in novel genes, not yet implicated in LCA and similar diseases. For this purpose, we use next generation or deep sequencing of retinal gene panels (NGS panels) and exome sequencing, respectively.
TEAM MEMBERS
Wolfgang Berger (PhD)
Silke Feil
Fatma Kivrak Pfiffner
Samuel Koller (PhD)