Arrhythmogenic Right Ventricular Cardiomyopathies (ARVCs)
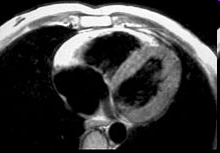
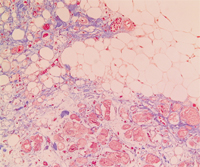
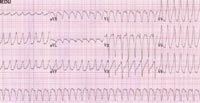
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a familial heart-muscle disease. It causes sudden death in young people and athletes. The disease is characterized by either massive or partial progressive replacement of the myocardium by fatty or fibro-fatty tissue. This infiltration provides a substrate for electrical instability and leads to life-threatening ventricular arrhythmias. ARVC accounts for up to 20% of sudden cardiac death in young people. The disease was first described in 1978. The prevalence of the disease in the general population has been estimated to range from 1 in 2000 to 1 in 10 000. Eighty percent of ARVC cases are diagnosed in patients with less than 40 years of age.
Genetic heterogeneity
In the last two decades the advances in molecular biology and genetics of ARVC have provided significant information in understanding the disease etiology. Interactions between mechanical disruption of cell-cell adhesion and defects of desmosomal-mediated intracellular signalling are likely to be involved in the pathogenesis of the ARVC phenotype.
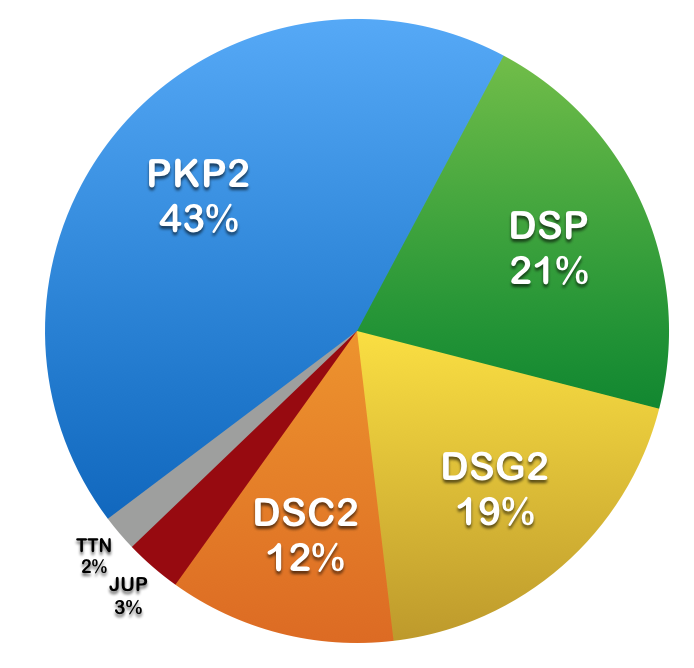
To date, at least 10 genes are associated with ARVC, 6 of which contain >95% of the reported mutations (n=482). However, in approximately 50% of the patients no mutation is detectable in one of the known genes. The discovery of the causative genes and mutations for ARVC offers the possibility of identifying genetically-affected individuals before a potentially malignant clinical phenotype occurs. Early detection of ARVC and preventive therapy of young individuals at highest risk of experiencing sudden cardiac death may be improved by molecular genetic screening within affected families and may alter the clinical management of patients.
Table: Summary of 6 most frequently mutated genes implicated in ARVC.
| Gene | Number of Exons | Protein | Number of Amino Acids | Number of Mutations* |
|---|---|---|---|---|
| PKP2 | 14 | Plakophilin-2 | 881 | > 400 |
| DSP | 24 | Desmoplakin | 2871 | > 150 |
| DSG2 | 15 | Desmoglein-2 | 1118 | > 150 |
| DSC2 | 17 | Desmocollin-2 | 901 | > 100 |
| JUP | 14 | Junction plakoglobin | 745 |
~ 20 |
| TTN | 363 | Titin | 35991 | ~ 15 |
* The numbers refer to disease-associated sequence variants listed in the Human Gene Mutation Database (HGMD) as of June 2022.
We focus on the identification of novel ARVC genes and mutations through next generation sequencing (NGS), including exome analysis in families with ARVC phenotype but no mutation in one of the known genes. We hypothesize that other mechanisms may be implicated in the ARVC pathophysiology, since about 50% of the cases with clear phenotype remain genetically elusive. Exome sequencing is defined as the selective sequencing of all exons in a genome to identify novel genes associated with rare and common diseases. Since most mutations causing monogenic disorders are exonic, this approach is especially promising for the analysis of monogenic diseases.
ARVCs: Diseases of the Desmosome
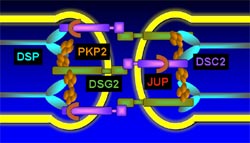
ARVC is understood as a disease of the desmosome, a cell structure specialized for cell-to cell adhesion. The primary role of the desmosome is to resist mechanichal stress; thus this junctions are particularly important in heart and skin tissues. They are also mediators of various signaling pathways, regulate the transcription of genes involved in adipogenesis and apoptosis and mantain proper electrical conductivity through regulation of gap junctions and calcium homeostasis. Desmoglein (DSG2 in green) and desmocolin (DSC2 in purple) are transmembrane proteins that bridge the space between adjecent cells, their cytoplasmic domains attach to desmoplakin (DSP in blue) via plakoglobin and plakophilin (JUP and PKP2 in red and orange).
TEAM MEMBERS
Wolfgang Berger (PhD)
Urs Graf (PhD)
COLLABORATORS (Clinic for Cardiology, University Hospital Zurich)
Thomas Lüscher (MD)
Argelia Medeiros-Domingo (MD/PhD, Clinic for Cardiology, Bern)
Firat Duru (MD)
Corinna Brunckhorst (MD)
Ardan Saguner (MD)